
The Clinical Data Interchange Standards Consortium (CDISC) creates standards that are now mandatory for a regulatory submission to the FDA and PMDA. Study Data Tabulation Model (SDTM) is one of the standards which provides a standard for streamlined data in collection, management, analysis and reporting. If your data is not using SDTM standards then a you will have to perform SDTM mapping to the latest version of SDTM standards as you prepare for your regulatory submission.
SDTM can support data aggregation and warehousing as well as data mining and reuse which are all important to statistical programming for the analysis of clinical trial data when attempting to prove the efficacy and safety of any Investigational New Drug Application (INDA). SDTM is also used in non-clinical data (SEND), medical devices and pharmacogenomics/genetics studies.
Within this blog we will cover the following topics on CDISC SDTM:
- Background on CDISC SDTM Standards
- The advantages of SDTM
- Latest CDISC SDTM Standards and Implementation Guides
- Medical Devices Implementation Guide
- Tobacco Implementation Guide v1.0
- Questionnaires, ratings, and scales (QRS)
- Therapeutic Areas
- Creating custom or non-standard CDISC SDTM domains
- Demonstrate SDTM mapping
- SDTM Automation
- SDTM Compliance
Background
CDISC has always recognized the need for the development of data standards to improve the process of electronic submissions and exchange of clinical trials information for the benefit of the pharmaceutical industry. SDTM, initially known as Submission Data Model (SDM), was developed by the CDISC Submission Data Standards (SDS) Team in 2004. The SDS Team is comprised of members from different sections of the pharmaceutical industry such as pharmaceutical companies and CROs. The SDS Team meets regularly to review SDTM standards, to continuously improve standards to meet industry requirements. These meetings are also attended by key members from the FDA. CDISC SDTM standards are rapidly evolving with the release of various Therapeutic Area (TA) specific standards as well as regulatory input and requirements.
SDTM Advantages
One interesting question that has come up in the past was, "If it is not yet compulsory to submit data in CDISC SDTM format then why should we change our standards to do this?". Although for FDA and PMDA submissions this is now a requirement, it is clear from listening to other people’s experiences why should we follow SDTM standards. Let’s go through some advantages of SDTM to better understand this.
Standarized Data
The main advantage of SDTM is to enable the industry to maintain and follow the same consistent standards for tabulation datasets across all studies. SDTM provides a uniform standard from study to study to ease data exchange internally and across vendors. The CDISC SDTM model is reviewed by many experts across the industry on an on-going basis to strive towards the best standard and to continuously improve the model. Having standardized data means that pharmaceutical companies should be able to collaborate on joint efforts more easily, and in the case of acquisitions, the clinical trials data from the company being acquired should be easy to integrate. The standards are also moving beyond the traditional drug reviews, also covering medical devices. Standardization between studies within a company will also provide more efficiency for the individual company, in terms of standardized code to produce domains and for reporting and analyses. This is an area that Quanticate is focusing on and it can save a lot of time in creation and validation of domains. Having a standardized structure will also make it easier to check the integrity of data from a data management perspective. Standardized checks could be written and performed while a study is still active. There are many such tools that have leveraged this over the years to perform validation, which we will cover later.
Review of Data
Another advantage of SDTM is in the actual review of the data. SDTM datasets follow standard dataset structures and variable attributes which makes it easier for regulatory bodies such as the FDA and PMDA to review data. Regulatory bodies have developed standardized tools for performing checks on submission data which significantly reduce review time and ensuring a basic level of quality before further review is undertaken. Reviewers can be trained on the data standards and standard software tools, so they are able to work more effectively with less preparation time. In fact, the FDA has their own reviewer tools that are run to obtain the information they require to review a submission. It is not necessary to provide subject listings with the submission which would result in fewer questions and faster drug approval time. Similar tools could be used internally by pharmaceutical companies and CROs to help with the review of safety and efficacy of study drugs. For example, standard tools can be developed which can create patient profiles. Hence, one could run a simple report to present the data for a subject (e.g. adverse events, concomitant medications or abnormal clinical laboratory results, electrocardiogram [ECG] or vital sign parameters) in such a way that is consistent, easy to interpret and could show whether the subject was receiving treatment when a particular adverse event or abnormal laboratory result occurred.

SDTM Implementation Guide: Human Clinical Trials
SDTM Implementation guide provides guidance on implementation of core SDTM standards and provides structure of various domains. If collected data is having relevant domain provided in the implementation guide, then this data should be mapped to the domain provided in the implementation guide. If collected data does not have relevant domain in the implementation guide then this data should be mapped to custom domains created based on classes defined in the core SDTM model.
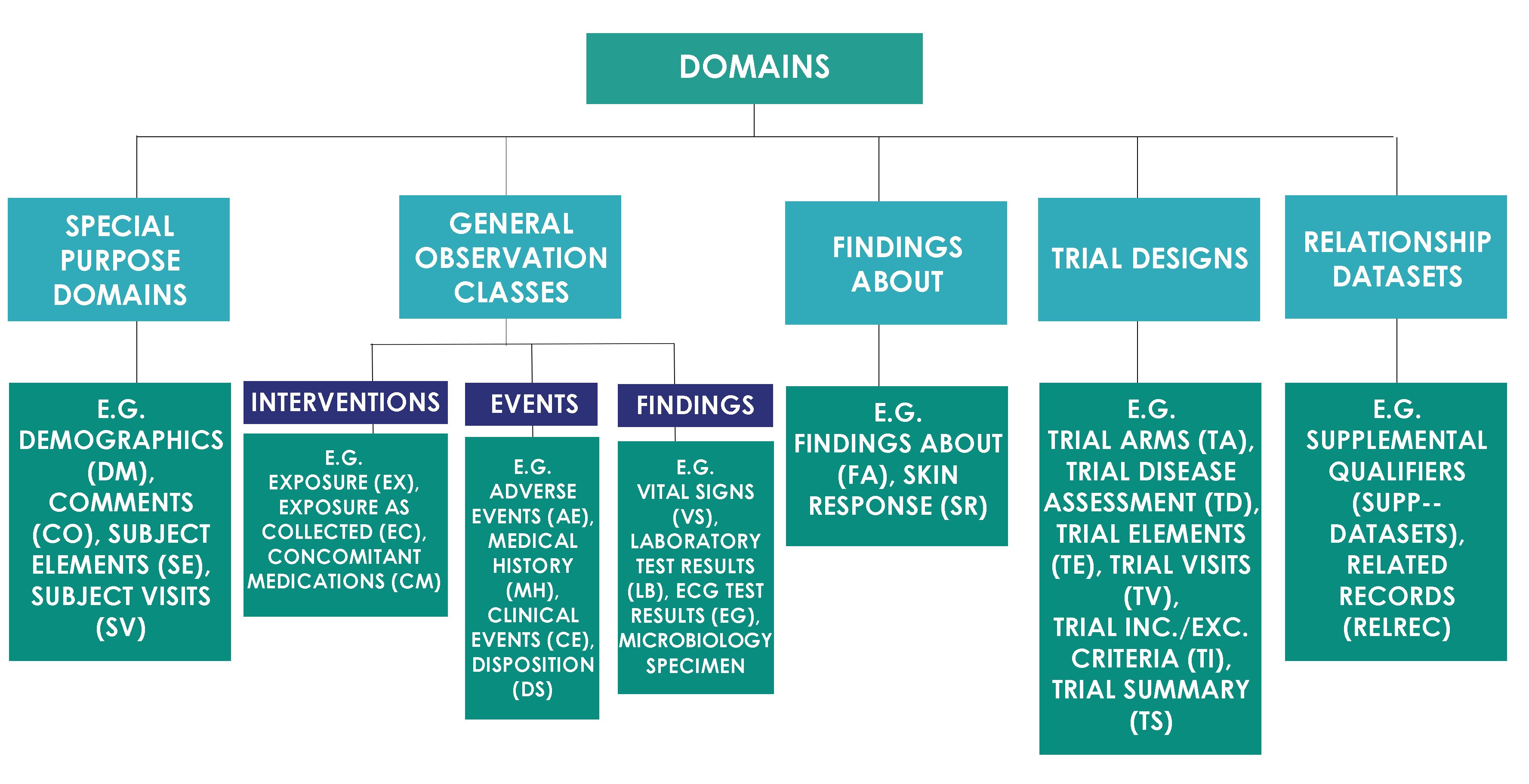
SDTM Domain Classes
SDTM domains are classified into the classes as shown in the image below.
Variable Role
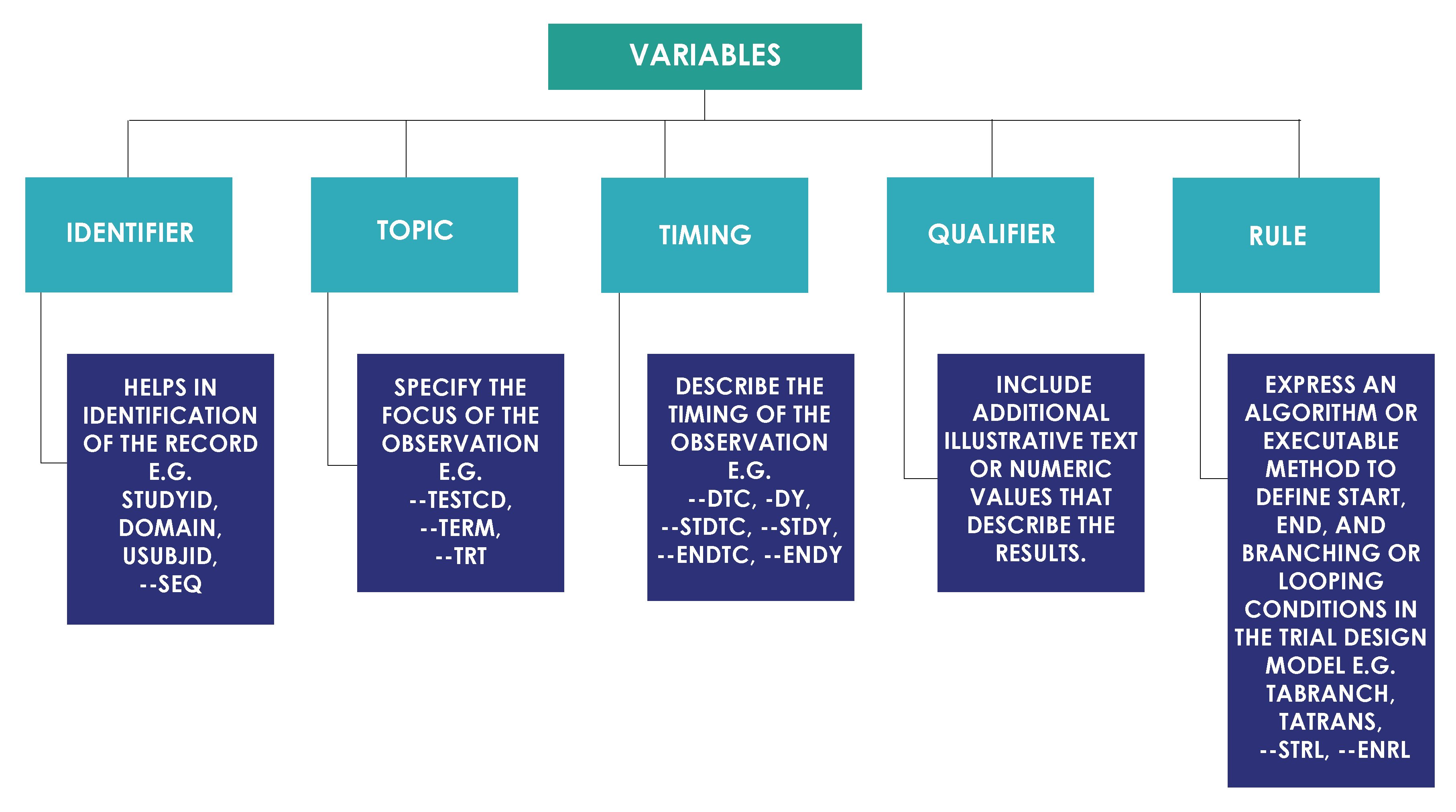
Each domain contains subject's data values collected during clinical trial that are organized as a table of observations (rows) and variables (columns). Each domain can have a list of potential variables. All variables based on their roles are classified into categories as shown in the image below.
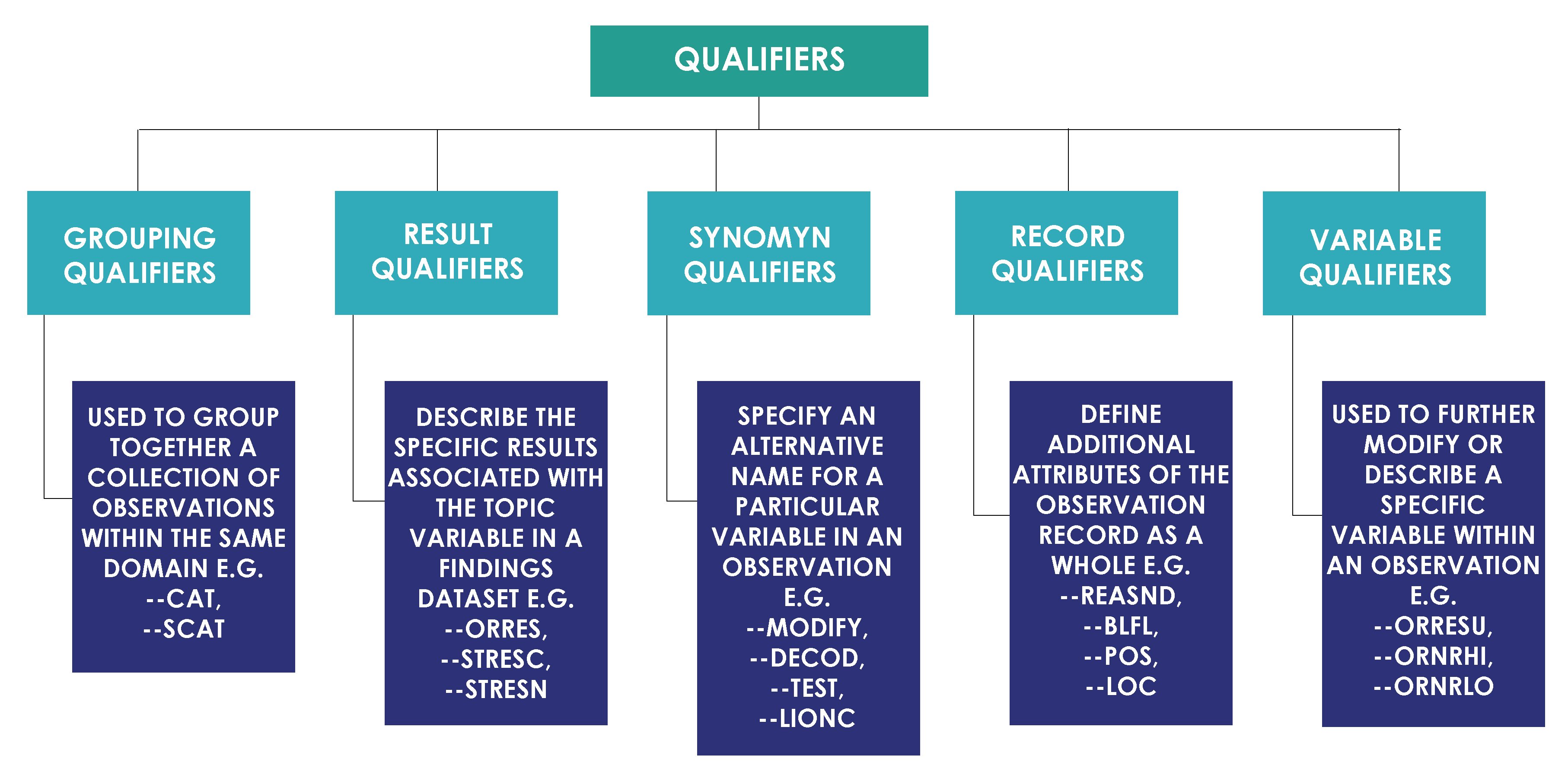
Qualifier variables based on their usage are further classified as shown in the image below.
CDISC Core Variables
There is a concept of core variables in the CDISC in order to control variables with missing values in the submissions. CDISC SDS Team categorizes SDTM variables as below.
- Required Variables: Required variables must always be included in the dataset and cannot be null for any record.
- Expected Variables: Expected variables may contain some null values, but in most cases will not contain null values for every record. When no data has been collected for an expected variable, however, a null column must still be included in the dataset, and a comment must be included in the define.xml to state that data was not collected.
- Permissible Variables: Permissible variable should be used in a domain as appropriate when collected or derived. The Sponsor can decide whether a Permissible variable should be included as a column when all values for that variable are null. The sponsor does not have the discretion to exclude permissible variables when they contain data.
There is a concept of core variables in the CDISC in order to control variables with missing values in the submissions. CDISC SDS Team categorizes SDTM variables as below:
- Required Variables: Required variables must always be included in the dataset and cannot be null for any record.
- Expected Variables: Expected variables may contain some null values, but in most cases will not contain null values for every record. When no data has been collected for an expected variable, however, a null column must still be included in the dataset, and a comment must be included in the define.xml to state that data was not collected.
- Permissible Variables: Permissible variable should be used in a domain as appropriate when collected or derived. The Sponsor can decide whether a Permissible variable should be included as a column when all values for that variable are null. The sponsor does not have the discretion to exclude permissible variables when they contain data.
SDTM Implementation Guide for Medical Devices
Medical devices are any instruments, apparatus or materials to be used in human for diagnosis, prevention and treatment of the disease. Devices are an important and growing part of the medical world, both on their own and in combination with drugs or biologic agents. Medical devices vary both in their intended use and indications for use e.g. low risk devices like tongue depressors, medical thermometers, bedpans, etc. and high-risk devices like implants, prostheses, etc. Medical Device Implementation Guide to the Study Data Tabulation Model defines recommended standards for the submission of data from clinical trials in which medical devices were used. This implementation guide is based on the original SDTM Implementation Guide (SDTMIG) developed for human clinical trials.
Domains per SDTMIG-MD are classified as shown in the image below.

Below image from the SDTMIG-MD explains the relationship of medical device domains with existing SDTMIG domains.

Device Identifiers (DI)
Device Identifiers (DI) is a Study Reference domain that provides a mechanism for using multiple identifiers to create a single identifier for each device. This domain provides primary device identifier variable (SPDEVID) for linking data across device domains. This is separated from the Device Properties (DO) domain because DI contains the total set of characteristics necessary for device identification, whereas DO contains information important for submission but that are not part of the device identifier.
| STUDYID | DOMAIN | SPDEVID | DIPARMCD | DIPARM | DIVAL |
| MD | DI | PC-001 | MANUF | Manufacturer | Ops Limited |
| MD | DI | PC-001 | MODEL | Model | 2000 |
| MD | DI | PC-001 | SERIAL | Serial Number | 001 |
| MD | DI | PC-001 | DEVTYPE | Type of Device | Pacemaker |
Device Properties (DO)
Device Properties (DO) is used to report characteristics of the device that are important to include in the submission and do not vary over the course of the study but are not used to identify the device e.g. shelf life, physical properties, etc.
| STUDYID | DOMAIN | USUBJID | SPDEVID | DOTESTCD | DOTEST | DOORRES | DORRESU |
| MD | DO | MD-999-001 | PC-001 | COMPOS | Composition | Titanium | |
| MD | DO | MD-999-001 | PC-001 | SHELLIFE | Shelf Life | 6 | Years |
| MD | DO | MD-999-001 | PC-001 | PWEIGHT | Pacemaker Weight | 50 | gms |
Device Exposure (DX)
The Device Exposure (DX) domain records the details of a subject’s direct interaction or contact with a medical device or the output from a medical device, usually but not always the device under study.
| STUDYID | USUBJID | SPDEVID | DXTRT | DXLOC | DXLAT | DXSTDTC | DXENDTC |
| MD | MD-999-001 | PC-001 | Pacemaker | ATRIUM | RIGHT | 2024-02-05 |
Device-In-Use (DU)
Device-In-Use (DU) contains the values of measurements and settings that are intentionally set on a device when it is used, and may vary from subject to subject or other target. These are characteristics that exist for the device, and have a specific setting for a use instance. DU is distinct from Device Properties (DO), which describes static characteristics of the device.
Device Tracking and Disposition (DT)
Device Tracking and Disposition (DT) represents a record of tracking events for a given device (e.g., initial shipment, deployment, return, destruction). Different events would be relevant to different types of devices. The last record represents the device disposition at the time of submission.
| STUDYID | DOMAIN | SPDEVID | DTTERM | DTPARTY | DTPRTYID | DTSTDTC |
| MD | DT | PC-001 | SHIPPED | SITE | 999 | 2024-02-02 |
| MD | DT | PC-001 | IMPLANTED | SUBJECT | 001 | 2024-02-05 |
Device Events (DE)
Device Events (DE) contains information about various kinds of device-related events, such as device malfunctions. A device event may or may not be associated with a subject or a visit. If a device event, such as a malfunction, results in an adverse event, then this information should be recorded in the Adverse Events (AE) domain. The relationship between the AE and a device malfunction in DE can be recorded using RELREC domain.
Device-Subject Relationships (DR)
Device-Subject Relationships (DR) links each subject to devices to which they have been exposed. This domain provides a single, consistent location to find the relationship between a subject and a device, regardless of the device or the domain in which subject-related data may have been collected or submitted.
| STUDYID | USUBJID | SPDEVID |
| MD | MD-999-001 | PC-001 |
Tobacco Implementation Guide v1.0
Tobacco Implementation Guide was prepared for the collection, tabulation, analysis, and exchange of tobacco product data such as:
• Cigarettes
• Electronic nicotine delivery systems (ENDS; vapes)
• Roll-your-own tobacco products
• Smokeless tobacco products
• Cigars
• Pipe tobacco products
• Waterpipe tobacco products
• Heated tobacco products (HTPs)
The TIG provides guidance for the:
- collection of study data with case report forms (CRFs) using the Clinical Data Acquisition Standards Harmonization (CDASH) model,
- tabulation of study data using the Study Data Tabulation Model (SDTM), and
- creation of analysis datasets using the Analysis Data Model (ADaM), with
- references to additional CDISC standards and resources to support implementation.
This guide has new datasets that can be used for displaying collected data. For example, Tobacco Product Identifiers and Descriptors (TO), Product Design Parameters (PD), Tobacco Product Testing (PT), Tobacco Ingredients (IT), Non-Tobacco Ingredients (IN), Ingredient Quantities by Component (IQ), Environmental Storage Conditions (ES).
Questionnaires, ratings and scales (QRS)
CDISC developed QRS supplements that provide information on how to structure the data. Standard mapping and examples can be used from the CDISC site (e.g. European Quality of Life Five Dimension Five Level Scale, European Quality of Life Five Dimension Three Level Scale, MS Quality of Life – 54, etc.) or it can be used as a guidance on how to map --CAT, -- TESTCD, --TESTCD.
CDISC creates supplements for four types of instruments:
- Questionnaires: Questionnaire instruments are stored in the Questionnaires (QS) domain and are named, standalone instruments designed to provide an assessment of a concept. Questionnaires often have a defined standard structure, format, and content; consist of conceptually related items that are typically scored; and usually document methods for administration and analysis. Questionnaires consist of defined questions with a defined set of potential answers. Most often, the primary purpose of questionnaires is to generate quantitative statistic to assess a qualitative concept.
- Functional Tests: Functional Test instruments are stored in the Functional Tests (FT) domain and are named, standalone task-based evaluations, designed to provide an assessment of mobility, dexterity, and/or cognitive ability. A Functional Test is not a subjective assessment of how the subject generally performs a task. Rather, it is an objective measurement of the performance of the task by the subject in a specific instance. Functional Tests have documented methods for administration and analysis and require a subject to perform specific activities that are evaluated and recorded. Most often, Functional Tests are direct, quantitative measurements.
- Clinical Classifications and Disease Response: Clinical Classifications and Disease Response instruments or criteria are represented in the Disease Response and Clin Classification (RS) domain.
source: https://www.cdisc.org/standards/foundational/qrs
Therapeutic Areas
Therapeutic Area User Guides (TAUGs) extend the Foundational Standards to represent data that pertains to specific disease areas. They provide disease-specific metadata, examples, and guidance for applying CDISC standards across various applications, including international regulatory submissions.
TAUGs are classified by Disease Areas as shown in the image below.

Creating Custom / Non-Standard Domains
When creating a custom domain, one should first confirm that there are no published domains available into which the data can be mapped. This can be done by checking against the reserved domain codes listed in the appendices of the current SDTM Implementation Guide, Medical Devices, Tobacco IG or by looking through a relevant Therapeutic Area User Guide if one is available for the indication under investigation. The following is not acceptable when creating custom domains:
- The nature of the data is the same as in another published domain.
- The custom domain is being created due to separation based on time.
- The data have been collected or are going to be used for different reasons. For example, if a lab parameter is collected for efficacy purposes the data must be represented in the LB domain and not in a custom ‘efficacy’ domain. The same applies to pharmacodynamics data that need to be PC and PP because the information comes from measurements of plasma serum.
- Data that were collected on separate CRF modules or pages and together may fit into an existing domain.
- It is necessary to represent relationships between data that are hierarchical in nature. Here, RELREC can be used instead.
Once it is confirmed that the data does not fit with any published domains, it should be determined which of the three general observation classes best fits the topic of the data since the custom domain must fit in to one of these. The next step is to determine a two-letter domain code for the custom domain. This should not be the same as the code for any published or planned domain. The domain codes X-, Y- and Z- are reserved for sponsor use, where the hyphen may be replaced by any letter or number. This domain code then will be the name of the domain and will also be used to replace all prefixes of variables from the class upon which it is based. The following steps can then be followed to create the custom domain:
- Select and include the required Identifier variables (STUDYID, DOMAIN, USUBJID and --SEQ) and any permissible Identifier variables (--GRPID, --REFID and --SPID).
- Include the Topic variable from the identified general observation class (--TRT for interventions, --TERM for events and --TESTCD for Findings).
- Select and include the relevant Qualifier variables from the identified general observation class only. These can be found in sections 2.2.1, 2.2.2 and 2.2.3 of the Study Data Tabulation Model (SDTM) before v2.0 or in sections 3.1.1, 3.1.2 and 3.1.3 of the SDTM v2.0 or later.
- Select and include the applicable Timing variables. These can be found in Section 2.2.5 of the Study Data Tabulation Model (SDTM) before v2.0 or in sections 3.1.5 of the SDTM v2.0 or later and relate to all general observation classes.
- Set the order of the variables within the domain: identifiers must be followed by topic variables, qualifiers and finally timing variables. The variables must then be ordered within these roles to match the order of variables given in sections 2.2.1, 2.2.2, 2.2.3, 2.2.4 and 2.2.5 of the Study Data Tabulation Model before v2.0 or in sections 3.1.1, 3.1.2, 3.1.3, 3.1.4 and 3.1.5 of the SDTM v2.0 or later. The variable order in the corresponding Define-xml data definitions file must also match the order within the domain.
- Adjust the labels of the variables only as appropriate to properly convey the meaning in the context of the data being submitted in the newly created domain. Use title case for all labels.
- Ensure that appropriate standard variables are being properly specified by comparing the use of variables in standard domains.
- Ensure that there are no sponsor-defined variables added to the domain. Any sponsor-defined variables should be in the corresponding Supplemental Qualifier dataset.
Variable attributes within the domain and Supplemental Qualifier datasets must conform to the SAS Version 5 transport file conventions. For example, variable names must be no longer than 8 characters, variables labels must be no longer than 40 characters and data value lengths must be no longer than 200 characters. Also, the transport file for any SDTM dataset should not exceed 5 GB in size or domains may need splitting to fulfil this requirement and the split documented in the Data Reviewer’s Guide that accompanies the submission.
SDTM Mapping
SDTM mapping is a process of converting raw dataset to SDTM domains. Mapping generally follows process as described below.
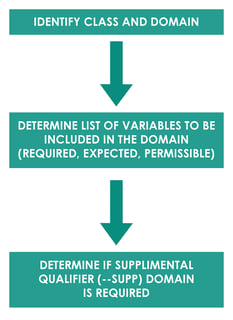
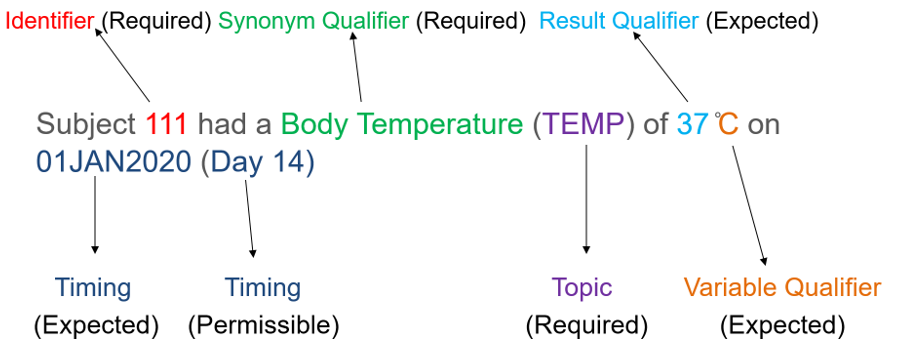
For example, Subject 111 had a Body Temperature (TEMP) of 37 ̊C on 01JAN2020 Day 14. Domain for this observation would be vital signs “VS”. The another Identifier variable is the subject identifier “111”. Topic variable value is the vital sign test code for body temperature “TEMP” and synonym qualifier value is “TEMPERATURE”. Result qualifier value is “37” and variable qualifier value for the result is “̊C”. Timing variable is the study date and day of assessment i.e. 01JAN2020 and Day 14. In this example, all the collected data fit into the standard SDTM variables so –SUPP domain is not required.
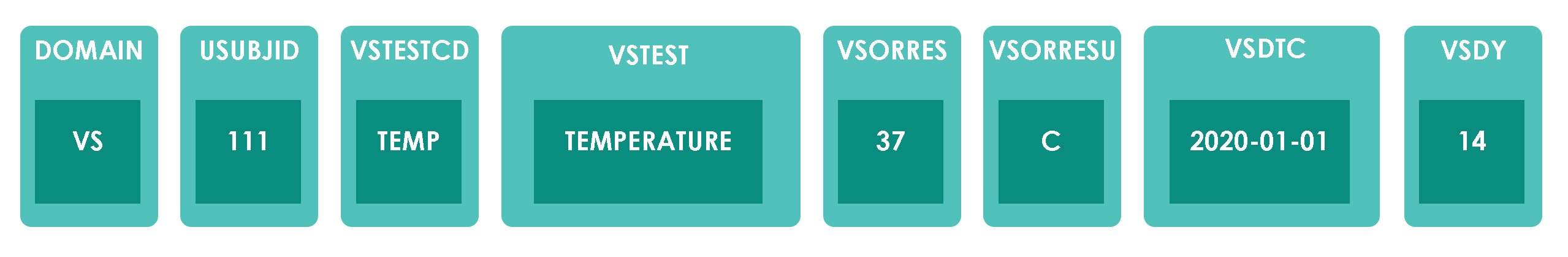
Based on splitting observation into variables, dataset structure would look like below. Data structure shown here is for example purpose only and does not contain all required and expected variables for VS domain.
Automation
Automation in SDTM programming is an important part of improving efficiency, accuracy, and consistency. By automating routine tasks and data transformations, programmers can significantly reduce the time and effort required for data preparation, ensuring that datasets are compliant with CDISC standards and spend more time on more complicated tasks. Automation minimizes the risk of human error, enhances data quality, and allows for faster and more reliable submission processes to regulatory authorities. Ultimately, it enables organizations to focus more on data analysis and interpretation, leading to better decision-making and outcomes.
SDTM Compliance
Compliance checks of CDISC SDTM data are also an important step in validating the conversion of raw datasets to SDTM datasets. Compliance checks of SDTM datasets can be done using open source validation tools like PROC CDISC in SAS, Pinnacle 21 Validator (previously known as OpenCDISC Validator) or WebSDM to name a few, but there are new tools being developed all of the time and thorough validation is seen as a clear objective to increase quality while reducing time for development and review cycles.
Another question that often appears is, "Is it possible to submit data as CDISC SDTM compliant if those data fail a particular requirement?" The data should follow CDISC SDTM standards unless there is a very good reason to deviate from the standard guidelines. However, it may not always be possible to follow standard guidelines exactly e.g. CDISC controlled terminology for adverse event severity expects AESEV variable to have values like MILD, MODERATE or SEVERE but if adverse event severity is collected on CRF as "Yes" or "No" and CRF updates are not possible then this does not conform to CDISC controlled terminology. Please note seriousness of adverse event is mapped to AESER and this example discusses about adverse event severity which is mapped to AESEV. When this SDTM is run thorough validation checks for CDISC compliance through some of the tools mentioned previously, this would result in an error. In such cases, it is acceptable to submit the data because it may just mean that a particular report for AESEV may not be able to run. However, detailed explanations of errors along with reasons for not being able to fix the errors should be added in the Study Data Reviewer's Guide (SDRG). However, as more companies and CRF designers become familiar with CDISC standards, this is now less of an issue than when CDISC was new to the industry. However, the fact remains that it is possible to submit data that fails specific checks, but this should be a last resort and a valid reason should be given in the SDRG with the submission.
Conclusion
The adoption of CDISC SDTM standards is now essential for regulatory submissions to the FDA and PMDA, underscoring their significance in the pharmaceutical and clinical research industries. These standards streamline data collection, management, analysis, and reporting, ensuring consistency and facilitating data exchange. By adhering to SDTM standards, organizations can enhance data integrity, improve collaboration, and expedite the review process, ultimately leading to more efficient drug development and regulatory approval. The comprehensive implementation guides and automation tools discussed in this article further support the practical application of these standards, reinforcing their role in advancing clinical programming and the successful submission of investigational new drug applications.

Related Blog Posts:
- Why do a 3rd of Regulatory Submissions fail the Technical Rejection Criteria?
- Using CDISC SDTM to Improve Cost and Quality in Integrated Summaries



.png?width=140&name=cdisc_gold_partner_80h%20(2).png)
