
The primary aims of Phase 1 clinical trials are to determine the safety, tolerability and pharmacokinetics (PK) of a compound. These are often performed on healthy volunteers, rather than participants who would benefit from the treatment, unless it is a therapeutic area where taking the treatment can make you ill, such as cancer or HIV. Trials have historically been conducted in the logical sequence of single ascending dose, multiple ascending dose, examination of preliminary effect of food on exposure, and potential drug-drug interaction, with assessments to determine the effect of gender, age, bioavailability and bioequivalence performed as necessary.
Additional studies may be performed, including definitive electrocardiogram (ECG) investigations to thoroughly evaluate the QT/QTc prolongation potential of a compound, which can increase the risk of potentially fatal proarrhythmias. This blog will describe the different designs commonly used in Phase 1 clinical trials.
The Different Phase 1 Trial Designs
Phase 1a - (Single Ascending Dose)
These are studies in which a small group of subjects (usually groups of three, although it is common for the first dose of a level to be given and assessed before dosing further subjects) receive a single dose of the compound in a clinical setting, usually a Clinical Research Unit (CRU). Close safety monitoring and usually PK assessments are performed for a predetermined time. If the compound is deemed to be well tolerated, and the PK data are broadly as expected, dose escalation occurs, either within the same group or a further group of healthy subjects, according to the approved protocol. Dose escalation usually continues until the maximum dose has been attained per the protocol unless predefined maximum exposure is reached, or intolerable side effects become apparent. Additionally, dose escalation may be discontinued (or may proceed more cautiously than planned) if there is evidence of a supra-proportional relationship between dose and exposure, such that exposures at higher dose levels become difficult to predict. This unpredictability means that the ability to monitor safety and tolerability within these trials is of paramount importance.
Study Design and Recent Advancements
Traditionally, phase 1a studies had followed simplistic designs, such as the 3+3 method. However, new methods, such as the BOIN method, allow for more flexible investigations (such as allowing for de-escalation and then re-escalating back to a level, or different DLT rates), which generates more information into the safety and tolerability of the dose levels, and a better chance of finding a more optimal Maximum Tolerated Dose (MTD) that previous methods allowed for (And therefore better success in later study phases).
Phase 1b (Multiple Ascending Dose)
These studies are conducted to investigate the PK and PD of multiple doses of the compound, again usually in a CRU. The dose levels and dosing intervals (i.e., time between consecutive doses) are selected as those that are predicted to be safe from single dose data. Samples are collected and analyzed to allow the determination of PK profiles and a better understanding of how the drug is processed by the body; with multiple dosing, a key part of the PK analysis is to identify if accumulation of the drug occurs.
As for phase 1a (single ascending dose) studies, dose escalation proceeds according to the protocol assuming strict safety and PK criteria are met. It is usual for 2-3 dose levels to be studied, at and above the expected therapeutic dose level(s) to determine the ‘safety margin’ for repeat dose administration.
It is not uncommon for a phase 2b study to also assess the Food effect (see below) at the same time.
Similarities and Differences between Phase 1a and 1b studies
There is an increasing trend for both the Phase 1a and 1b study to be contained in the same study protocol (often called a “dose-escalation and -expansion” study and split into stages within the protocol (stages A and B, for example), with language set to carry forward the information from stage A to stage B, thereby saving on time between stages preparing a new study. There is also the option to conduct a phase 1b/2a trial, with the same intention of accelerating the research process and combining elements of the study set-up procedure to save time, cost and resources.
Both Phase 1a and 1b studies have a high emphasis on safety. In both cases, it is likely the first time participants will be exposed to the treatments they get (either dose level, or multiple doses of a level). They are normally both conducted in similar settings to allow close monitoring for any potential reactions.
Phase 1a studies will only give participants a single dose (or cycle) of a treatment, whereas phase 1b will generally consist of multiple doses (or cycles) over a longer period of time. This allows for multiple measurements/assessments to be conducted in a phase 1b to see how the treatment works over a longer period (for example, whether absorption changes after multiple doses/cycles in a drug trial).

Food effect
Food effect studies are conducted to allow a preliminary assessment of how absorption of the compound is affected when administered after a specifically designed and standardized test meal, usually a high fat breakfast in the first instance. Such studies are usually conducted following a crossover design such that the same subjects receive doses of the study drug on different occasions under fasted and fed conditions. In such studies, each subject acts as his or her own control such that the impact of intersubject variability is reduced. These studies are important as the results are likely to contribute to the wording on the label as to when drugs should be administered relative to food in order to increase, or indeed decrease, absorption. Please see the table below as an example of potential wording and grouping.
|
|
Period 1 |
Period 2 |
|
Group A |
Take dose after consuming standardized test meal |
Take dose while fasted |
|
Group B |
Take dose while fasted |
Take dose after consuming standardized test meal |
To reduce the costs of drug development and time to market, the designs above have more recently been combined into single flexible protocols that include adequate dosing intervals to allow for the review of safety and/or PK data prior to study progression.
Studies Performed Subsequently
Drug-drug interaction
These studies form an extension of the in vitro and in vivo preclinical studies in which the routes of metabolism are identified. Correlation of the results allows the identification of enzymes which may be subject to inhibition or induction by the test drug or which are responsible for its metabolism. Clearly, the selection of drugs for drug-drug interaction studies is based upon those for which co administration is likely for the particular indication and target population of the compound. A common study design involves co administration of a compound with a strong inhibitor (e.g., ketoconazole) or a strong inducer (e.g., rifampicin) of the metabolizing enzyme cytochrome P450 3A4. The route of administration should be based upon that planned for the marketed compound, assuming this is known, and studies should use the highest proposed or approved doses of the test drugs and the shortest dosing intervals to maximize the likelihood of an interaction, unless this is precluded for safety reasons. There should be a particular emphasis on monitoring for adverse events (AEs) that occur at a greater frequency or severity during combination treatment than during treatment with either drug alone. In addition, there should be extensive PK sampling to allow for full characterization of the parent drug and important metabolites, if applicable, and any potential temporal relationship to AEs.
Bioavailability and bioequivalence
Studies to measure bioavailability and/or establish bioequivalence of a compound are important elements in support of regulatory submissions. For orally administered compounds, bioavailability studies elucidate the process by which a drug is released from the oral dosage form and moves to the site of action within the body. Bioavailability data provide an estimate of the fraction of drug absorbed, as well as its subsequent distribution and elimination. This can either be absolute bioavailability (the compound is compared to intravenous administration, which is assumed to be 100% bioavailable) or relative bioavailability (the compound is compared to another formulation or non-intravenous route of administration).
In bioequivalence studies, the systemic exposure profile of a test compound is compared to that of a reference drug product. To be considered bioequivalent (or statistically ‘non-different’), the active drug ingredient or active moiety in the test compound must exhibit a rate and extent of absorption that is comparable to the reference drug product, according to strict statistical rules. Two treatments are considered to be "not different" to one another if the 90% confidence interval (CI) for the ratio of the log transformed exposure measure falls completely within the range 80%-125%. If the 90% CI falls outside the 80%-125% range, the 2 treatments are concluded to be different from one another. Bioequivalence studies are performed if there is a change in dose form, structure, manufacturing process, or excipients to ensure the ‘new’ form is equivalent to the previous version. Bioequivalence studies are also performed when generic drugs are developed which contain the same active ingredients as the original formulation, which is no longer under patent protection. Bioavailability and bioequivalence studies are routinely conducted in a small number of subjects in Phase 1 CRUs and reported according to strict regulations.

Other Studies That may be Performed as Part of Phase 1 Studies
The absorption, distribution, metabolism and/or excretion (ADME) of a compound or its metabolites may be influenced by many factors including age, gender, genetics or social factors, such as smoking status. Phase 1 studies are not only conducted at the initial stages of drug development in man; whilst such studies are undoubtedly more common, additional Phase 1 studies are often conducted later in the development of a compound, alongside large Phase 3 studies in the target population/indication. The aim of these studies is to obtain data to support the wording for the label and may be the result of requests from regulatory authorities to further substantiate claims or address perceived weaknesses in a drug submission.
Traditionally, Phase 1 studies have not routinely been performed in the pediatric population. The US Pediatric Research Equity Act (PREA) 2003 (amended 2007) was signed into law to address the lack of pediatric information in drug product labelling. The PREA requires all applications (or supplements to an application) for a new active ingredient, new indication, new dosage form, new dosing regimen, or new route of administration to contain a pediatric assessment unless the applicant has obtained the appropriate waiver or deferral.
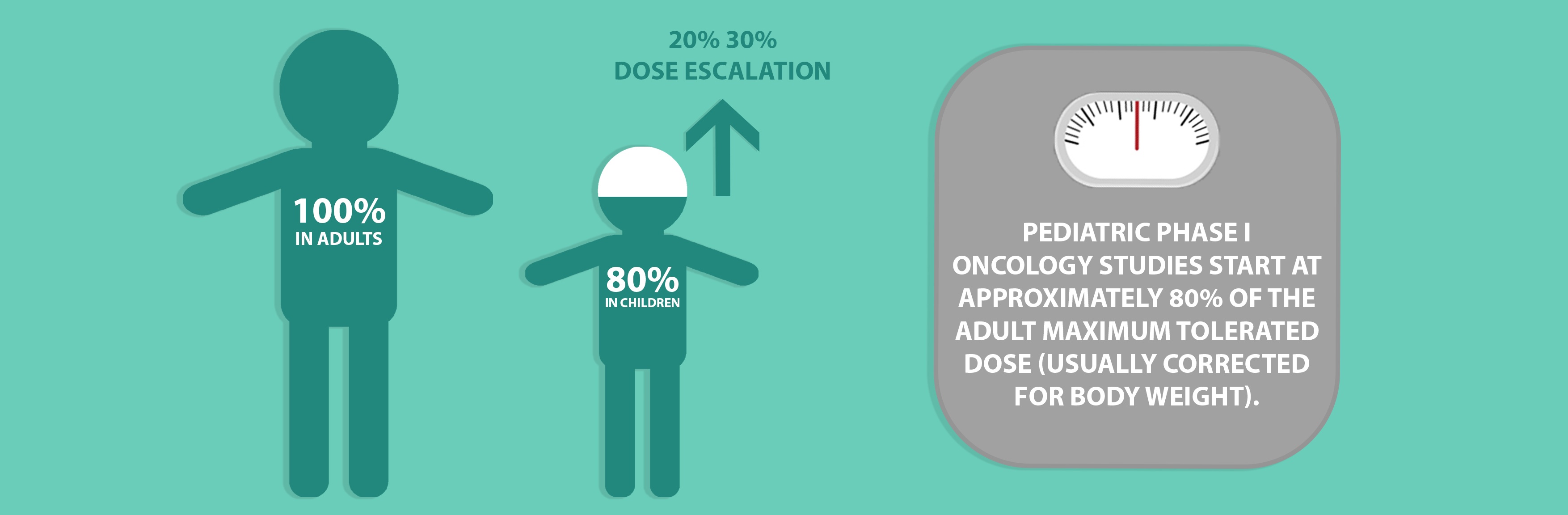
For therapeutic areas such as oncology, Phase 1 studies are seen as an important step in the evaluation of novel compounds. Such studies must include a potentially active treatment and must not be performed to only evaluate toxicity. Where performed, pediatric Phase 1 oncology trials are almost always conducted following adult Phase 1 studies and start at approximately 80% of the adult maximum tolerated dose (usually corrected for body weight), greatly diminishing the likelihood that a pediatric patient would be enrolled at a biologically ineffective dose. Dose escalation typically occurs in 20% 30% increments in successive cohorts and there is generally no intrasubject dose escalation. Studies are based on the assumption that children have a similar or higher threshold for toxicity than adults and the number of children required for Phase 1 studies are kept as low as possible.

Economical Designs for Phase 1 Studies
From a cost prospective the design of a phase 1 study can be positively influenced with proper planning, intelligent study design, and well thought out standardization of CRF design and statistical programming. With protocols that are written flexibly, many objectives can be addressed under one protocol (often with many parts) which can improve efficiencies in the drug development process and speed up the time to becoming Phase II ready.
Apart from combination studies, use of novel statistical designs can be used to improve Phase I studies or other clinical pharmacology studies in other ways. For example, the use of incomplete block designs (where each subject receives a subset of the treatments under study) can enable more treatments to be tested within a cross-over study than might otherwise be thought practical. This may arise in situations where different formulations of the same compound are to be evaluated.
Factorial designs can also be used to help explore several factors simultaneously in one study (e.g. for ascertaining the optimal dose levels for a combination product). For compounds where the expected variability of the pharmacokinetic parameters is high, resulting in a large sample size, the use of interim analysis or sequential designs can offer the opportunity to complete the study more quickly with fewer subjects, if the primary objective has been reached or the study looks as if it will fail to achieve it.

The place of Phase 1 trials in the drug development process is further outlined in the blog entitled, 'The Different Phases of Clinical Trials'.
To learn more about how our statistical consultancy team and medical writing services could support your phase 1 study designs please request a consultation below and a member of our team will be in touch with you shortly.



