
What is an ISS and an ISE?
The integrated summary of safety (ISS) and integrated summary of effectiveness (ISE) are critical documents required for regulatory submissions in the approval of new medicines/drugs or devices. These documents combine the safety and efficacy results from different clinical trials conducted for the investigational product and need to contain detailed analyses of all relevant data from the individual clinical study reports. ISS and ISE allow reviewers to compare results for individual endpoints across studies for the investigational product. Overall, ISS and ISE enable broad views of the investigational product’s overall efficacy and safety profiles.
Why Perform an ISS and ISE?
ISS and ISE are critical for regulatory submissions when combining trial results. They will feature in New Drug Applications (NDAs) which are required for the NDA submission to the United States Food and Drug Administration (FDA)[1]. However, ISS and ISE are not a regulatory requirement for the marketing authorization application (MAA) submission to EMA in Europe or to the Ministry of Health, Labour and Welfare (MHLW) of Japan. MHLW and EMA submissions will contain elements of ISS and ISE because they are part of the common technical document (CTD) format which must be followed for these regulatory submissions[2]. The Canadian regulatory authorities, Health Canada, made the Common Technical Document (CTD) format mandatory for regulatory submissions including NDAs on 1st January 2018 and is referred to as New Drug Submissions (NDS)[3].
The ISS and ISE sections within a CTD require a comprehensive amount of data analysis at the clinical trial reporting stage, conducted by statistical programmers. This analysis and supporting information is heavily formatted by medical writers to present the results within the CTD. Although there are no corresponding regulations requiring an ISE or ISS for biologic license applications (BLAs) submissions, applicants are encouraged to provide these analyses[4].
As well as a regulatory requirement, integrated summaries allow us to:
- Make informed decisions on an investigational drug’s efficacy and safety by combining multiple studies, instead of using data from single study to support evidence for drug’s efficacy and safety.
- Combining studies for analysis provides more robust statistical conclusions.
- See a larger pool of data relating to the investigational drug and better understand any variations in study results and the risks and benefits of these, as well as any unexpected trends.
- Discover data anomalies and improve data integrity when combined with centralized statistical monitoring technologies.
Why are Common Technical Documents (CTD) Important?
In general, for any regulatory submission, CTD is a common globally accepted format for the dossiers that are submitted to gain approval for medicinal products. The aim of creating the CTD was to provide a uniform format for drug approval and is the required format for all marketing authorization applications/new drug applications in the US, EU and Japan, Canada, and Australia.
A CTD is comprised by 5 modules, with 4 common global modules (2-5), and module 1 being specific to the region that you are submitting your CTD to.
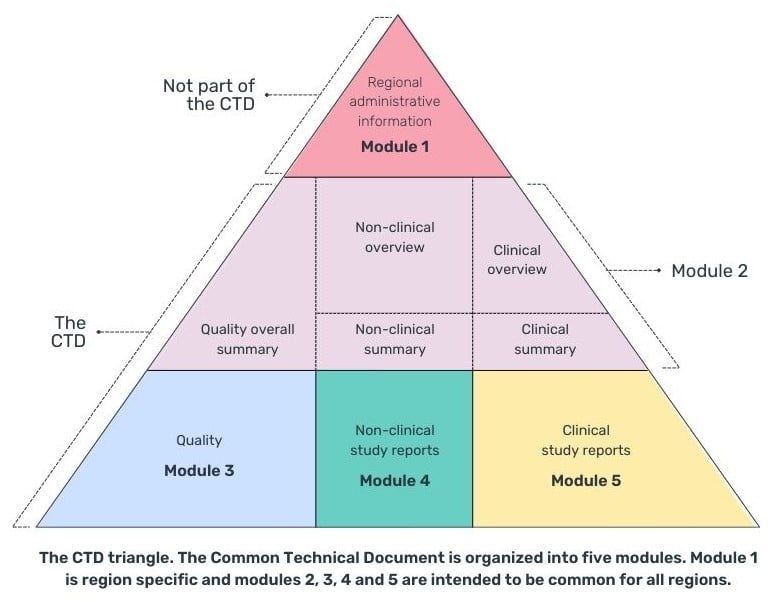
The summary of clinical efficacy (module 2.7.3) and summary of clinical safety (module 2.7.4) can be placed in module 2 (replacing the occurrence of integrated summaries in module 5.3.5.3) or in module 5.3.5.3 of module 5, the clinical study report (Table 1). However, it is more common for the integrated summaries to appear in module 5 as module 2.7 has a 400-page limit and unless it is a small study and comparison of studies, it is likely not to be enough space and module 5.3.5.3 would provide enough space to document the required analysis and appendices of tables, listings and figures.
Table 1: ISE- and ISS-Related Sections with Corresponding Regulations[4]
|
CTD Section |
US Regulation |
Remark |
|
2.5 Clinical Overview (~30 pages) 2.5.4 Overview of Efficacy 2.5.5 Overview of Safety |
NA |
Not a U.S. requirement, but recommended by ICH M4E |
|
2.7 Clinical Summary (~50 – 400 pages) 2.7.3 Summary of Clinical Efficacy 2.7.4 Summary of Clinical Safety |
21 CFR 314.50(c) (2)(viii) |
U.S. requirement for a clinical summary |
|
5.3 Clinical Study Reports 5.3.5.3 Reports of Analyses of Data from More than One Study (Including Any Formal Integrated Analyses, Meta- Analyses, and Bridging Analyses) |
21 CFR 314.50(d)(5)(v) 21 CFR 314.50(d)(5)(vi)
|
Integrated Summary of Effectiveness Integrated Summary of Safety |
(Source: Guidance for Industry (fda.gov) – https://www.fda.gov/media/75783/download p3.)
As a global standard, to all regulatory bodies, CTD guidance[2] is set and reviewed by the International Conference of Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH).
What to Include Within an ISS and ISE?
An integrated summary should not just be considered a summary of the details of individual study results held in the Clinical Study Reports (CSR), but rather, is a comprehensive and in-depth analysis of aggregated results used to make informed decisions. This analysis involves a synthesis of the results of individual studies, in an appropriate manner, to provide evidence of the safety and effectiveness of the drug. The integrated summary is far more than just a ‘summary’, detailing pooled analyses and discussing them in detail.
The main section of the ISS and ISE uses text to explain the study results, within this body there are tables and figures of important endpoint analysis. Finally, details of statistical approaches and listing, should be included in an appendix, which can also contain any additional supportive tables and figures. Let’s look at what to include in the ISE and ISS in more detail:
What to include in a Integrated Summary of Effectiveness
An ISE is used to define and support the effectiveness of the investigational drug’s effectiveness in terms of the endpoints and treatment aims as specified within the trials protocol. The ISE will include all available information on this effectiveness for each target claim, indication and recommended use. Below are its key components[5]:
- An integrated summary of the data demonstrating substantial evidence of effectiveness for each claimed indication.
- Evidence that supports the dosage and administration section of the labelling, including support for the recommended dosage and dose interval.
- Effectiveness data analyzed by sex, age, and racial subgroups, identifying any modifications of dosing for specific subgroups, including evidence to support the individualization of these subgroups. This should also include a note if specific subgroup(s) have been excluded from the trial.
- Effectiveness data from other subgroups of the population of patients treated, including any varied dosages and individualization based on these subgroup characteristics.
- Overview of the results, showing that they satisfy the regulatory requirements for approval, representing adequate and well-controlled studies demonstrating the claimed effect, especially if results are inconsistent or marginal.
- Examination of study-to-study differences in results, effects in subsets of the treated population, dose-response information from all sources, any available comparisons with alternative drugs, and any other information
- Detailed integrated analyses of relevant sources of information concerning effectiveness, including:
- Comparisons of individual studies to understand overall results and consistency.
- Comparison of results across multiple studies
- Pooled analyses for insights into the drug’s effectiveness across demographic and other subpopulations, dose-response relationships, onset, and duration of effect.
- Comparisons with alternative drugs, providing a context for the drug's effectiveness in relation to other available treatments.
- A list of individual studies with a brief description of the study design and results.
- Justification for any reliance on a single study.
- Analysis of evidence of long-term effects, tolerance, and withdrawal.
- Review of results from uncontrolled studies, if relevant in supporting claims of effectiveness.
This list is generated from more detailed regulatory guidance on what to include in an ISE.
What to include in a Integrated Summary of Safety
An ISS is offers the overall analysis and summary of the safety data of the investigational drug. Below are its core components[6]:
- A summary of the safety profiles from all clinical studies (can include Phase I studies in healthy volunteers; normally presented separately from study patients)
- The overall extent of exposure – the number of patients exposed (by gender and other demographic subgroups) and number exposed to various doses to the active drug for defined periods
- Demographic and other characteristics of study population – age, sex, ethnicity, body weight, primary & secondary diagnosis, concomitant therapy taken during study and other relevant prognostic variables such as smoking status and alcohol use
- Adverse events:
- Overall analysis of adverse events including occurrence rates, deaths, adverse dropouts and other potentially serious events in all studies
- Laboratory assessments
- Summary of animal data important to human safety
- Analysis of adverse effects relating to dose-response information– This should include any evidence of dose-response variations relating to age, sex or any other subpopulations.
- Drug-Drug interactions, acknowledging that a study of all is impossible but potential interactions, drugs likely to be co-administered with the new study should be identified, any unwanted effects and supportive pharmacokinetic and pharmacodynamic data
- Drug-demographic, and drug-disease interactions including pharmacokinetic (PK) and pharmacodynamic (PD) studies with an emphasis on diseases or demographic features that could alter metabolism or distribution of the drug
- Any other pharmacologic properties – especially those that have proved often to be pertinent to the use of drugs, including effects on Liver, Kidneys, heart, central nervous system (CNS) etc.
- Long-term adverse effects (6 months or more) and withdrawal effects
This list is paraphrased from Section H of more detailed original regulatory guidance on ISS.
What are the Challenges When Creating an ISS and ISE?
There are many challenges when producing an ISS/ISE, but most of them stem from trying to combine multiple studies that are independent of each other, with potentially different assessment schedules, endpoints, dosing regimens and data standards.
Similar to a single study, many of these issues can be mitigated, or avoided entirely, through proper planning and strong communication between the vendor and the sponsor.
An ISS/ISE Statistical Analysis Plan (SAP) is the key starting point to ensure that an ISS or ISE goes smoothly and produces a robust analysis that supports the regulatory submission that it is part of. There is a risk that a poorly planned integration will produce summaries that are not truly pooled, but just a side-by-side concatenation of the individual study tables.
Communication between sponsor and CRO are vital for ensuring a smooth pooling process. Some contributing studies may still be ongoing and proper understanding of the data availability/cleaning status will ensure that accurate timelines can be planned for the final pooling. It is also beneficial for all parties to have an understanding of the messaging in the submission and the intended labelling information that the sponsor is hoping to get approved.
These challenges are discussed in more detail below.
How to Plan an Effective ISS ISE/Integrated Summaries
An ISS ISE need to be designed and planned carefully in advance to ensure informed decision-making and effectiveness. A focus on the approval and whole lifecycle of the product, and not just the submission, will influence the quality and direction of the content. Traceability is key in all respects of the creation of information, from data that leads to knowledgeable decisions and the ultimate wisdom that forms the label of a product.
Approvability of a product depends, of course, on the data that is selected for collection during the planning of clinical trials. However, considerable resources, time and money can be saved by planning carefully how the summaries are structured to help make effective decisions and deliver clear messages supporting the target product claims.
The following questions need to considered when planning your integrated summary:
- Will this strategy for creating the CTD be discussed with authorities and will briefing documents be needed?
- What studies or populations are needed to fully support the proposed label?
- To what degree will you be re-using information and can this be maximized for future submission if you are planning several?
- How are you going to manage the traceability of the information from the collected raw data through to the different derived summaries?
- Have you ensured that your metadata from derivations of values is effectively management for later use in the submission and regulatory defense?
Analysis planning of these summaries may be started alongside the preparation of the Phase II studies to ensure appropriate endpoints, time points and patient populations are being considered to allow for pooling at a later timepoint.
Statistical Analysis Plan (SAP)
An integrated statistical analysis plan (SAP) for Safety or Efficacy which will include details of each study to be pooled for the ISS/ISE is important to an ISS/ISE plan. The SAP should include a list of integrated analysis tables, listings, and figure (TLF) outputs, in addition to mock-ups before the final TLFs. Once the scope of the integrated analysis is clear and supporting documents (electronic case report forms [CRFs], datasets specifications, etc.) are available for each individual study, programmers can start to plan and design integration datasets.
The SAP will also provide details of how data differences between the contributing studies will be handled, whether they are differences in data collection or the algorithms used for reporting each study. This ensures that the combined analyses are robust and meaningful.
Meeting Sponsor Expectations
Guidelines from agencies on the preparation of integrated summaries for regulatory submissions are often lacking in terms of providing specific direction on content. What is important to sponsors at the point of submission can vary, but it is crucial to meet the needs of the sponsor, understand the details and then allocate the appropriate time and set a budget for thorough ISS and ISE that will be certain of meeting the regulator’s expectations.
Visualization Output Templates
Visualization of the summaries in terms of creation of output templates is defined at this early stage. These will undergo multiple reviews and updates as the knowledge of the drug increases and as the process becomes more focused, with data becoming available from early phase studies.
Communication and Collaboration Between Cross Functional Teams
Forming a well-integrated and closely-knit team, including the programmer, statistician, medical writer, and physician, is key to ensuring that reviews are performed early enough to allow for a timely setup and reporting. This close partnership, especially between the programmer and statistician, fosters a clearer understanding and enables the delivery of high-quality documents, especially when working on highly technical pieces such as the CTD, all within tight timelines.
Data Storage
Early planning is crucial in determining how data, information, and knowledge are stored. During a product’s lifecycle the data points collected from clinical studies can be used in endless ways for information creation and decision-making. By establishing a system for easy traceability and using effective tools to manage it, you can clearly understand the basis of data-driven decisions and effectively respond to regulatory defense queries. This careful planning and documentation of each piece of data, which form the cornerstone of your product’s label for safety, efficacy, and targeted populations, are vital. These elements, integral to your organization’s intellectual property, must be meticulously planned and documented through metadata and shared with consideration, underpinning the long-term benefits to your product's label.
Creation of Integrated Summaries
There are many considerations in the actual creation of the integrated summaries. These should be laid out in the ISS/ISE SAP, but it is vital that the sponsor is fully engaged with these decision points as they will inevitably affect the messages that can be pulled from the pooled data.
Dictionary Versioning
Dictionary versions (MedDRA, WHODRUG, etc…) should be up-versioned to the latest version used within the pooled studies to ensure that adverse events and con meds can be pooled and summarized successfully. This process will need a review to see that any adverse events of special interest are not re-coded, or if they are, that this can be accounted for in the analyses.
Harmonizing of Variables
There is no guarantee that common variables between the pooled studies will contain the same information. This is particularly the case with CDISC studies, where standard variables – e.g. ANLxxFL, AVALCATy – may have completely different uses in each study. These flags will need careful mapping to ensure that definitions are maintained correctly in the pool.
Programming Logic
Each contributing study will have its own SAP and while many algorithms are standard within the industry, there may be subtle differences in some endpoints. These may need to be harmonized fully, with rederivation of some variables, or they may simply need accounting for in the analyses. Statisticians can advise on what is necessary to ensure a robust analysis.
CDISC Compliance
Contributing studies may or may not have been delivered to CDISC standards, and if they were then it is likely that they were delivered to different versions of the implementation guides. A key focus of a good integrated summary package is ensuring that the pooled data is fully CDISC compliant to the latest standards, to ensure that there are no roadblocks during the submission process.
What is the best approach to Review Integrated Summaries?
While there is often pressure to rush the submission process, it is important to balance this with the need for careful review. Project teams should have reviewed the output tables using blinded and unblinded data to make sure that they have all the graphs and summary tables required to best interpret the information. Depending on the company/supplier setup, coordination of the review cycle will generally be done by the clinical data service provider. Getting the input of medical writers and physicians before the unblinding of the Phase III studies is vital but difficult to achieve. Planning review meetings early in people’s calendars and making sure the relevant personnel have performed a quality review prior to the meeting to enable a productive review can be critical in saving on re-engineering of the project’s critical path.
A thorough and careful final review not only impacts the speed at which submissions are reviewed and products are introduced to the market but also affects the quality of the label that is ultimately approved. A well-prepared submission, with information that is well organized, traceable, and clearly understood, is more likely to facilitate a smoother regulatory defence, potentially reducing the number of questions from authorities and the time required for responses. The primary goal should always be the approval of the product, achieved through a high-quality submission that require only one review cycle. The ultimate measure of a quality submission lies in its efficiency in obtaining approval, characterized by fewer queries and a faster turnaround time.
Pre-empt Potential Regulatory Questions
It is important to pre-empt any questions that may come from regulatory authorities to improve the review time of a product and enhance the long-term reputation of your organization. While most project teams believe they have supplied every eventuality in terms of summary tables, insights can always be gained from other submitted projects. Forming a team of experts to critically review the summaries developed for your submission can be an effective strategy. This team should aim to anticipate and address potential questions that regulatory reviews potentially might have.
Conclusion
Effective planning of your integrated summaries should start early in the process of the approval of a product. At Phase II, you should start to plan and be prepared to adapt the plan as knowledge of the product increases. Communicate effectively with the relevant individuals in your teams and ensure close partnerships throughout between your programmers and statisticians. Using metadata, define the standards of how the data is captured, how information is derived and stored, and how knowledge is acquired and subsequently used upfront. This will pay dividends in the traceability of the crown jewels of your product in the long run. Don’t rush the end game and be ready for any questions that may come your way.
Ultimately, an optimal clinical program with great vision, design, and strategy will provide you with the expected results to support your label, but this does need to be backed-up with the right summaries to best explain and interpret your hard work. You may have the right data to strengthen your target product claims, but unless the correct information and knowledge is generated to support this, it could take you a lot longer to get to the approval stage if your planning and summaries are not thought through. The summaries must communicate the vision defined through its selective program design and expert interpretation of the quality data generated, ensuring a coherent and compelling narrative that aligns with regulatory expectations and enhances the likelihood of a successful product launch.
At Quanticate we have a wealth of experience in producing integrated summaries for our clients with our ISS ISE services. Working collaboratively across our biostatistics, programming and medical writing departments we can support you with a successful regulatory submission and product launch when you need to combine multiple studies to prove your drugs efficacy and safety. For more information, please submit a request for consultation below.

Related Blog Posts
References
[1] FDA Center for Drug and Biologics Evaluation and Research (CDER and CBER). (April 2009). Guidance for Industry: Integrated Summaries of Effectiveness and Safety: Location Within the Common Technical Document. Retrieved from https://www.fda.gov/downloads/drugs/guidances/ucm136174.pdf
[2] (2018). CTD: M4: The Common Technical Document. [online] Retrieved from https://www.ich.org/page/ctd
[3] ca. (2018). Updated Notice – Mandatory use of the Electronic Common Technical Document (eCTD) format – Canada.ca. [online] Retrieved from https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/announcements/notice-mandatory-requirements-using-common-electronic-submissions-gateway-technical-document-format.html
[4] FDA Guidance for Industry – Integrated Summaries of Effectiveness and Safety: Location Within the Common Technical Document. April 2009. https://www.fda.gov/media/75783/download
[5] FDA Center for Drug and Biologics Evaluation and Research (CDER and CBER). Integrated Summary of Effectiveness: Guidance for Industry. October 2015. Retrieved from https://www.fda.gov/downloads/drugs/guidances/ucm079803.pdf
[6] Food and Drug Administration. (1988). Guideline for the format and content of the clinical and statistical sections of new drug applications. FDA, US Department of Health and Human Services Rockville, MA, USA. Retrieved from https://www.fda.gov/media/71436/download / https://www.fda.gov/regulatory-information/search-fda-guidance-documents/format-and-content-clinical-and-statistical-sections-application



.png?width=140&name=cdisc_gold_partner_80h%20(2).png)
