Traceability of data is a key concept that is becoming increasingly important to Pharmaceutical, Biotechnology and medical device companies in a rapidly evolving regulatory environment. This blog aims to discuss the concept of traceability of clinical information, consider why it is so important and how it may be achieved. Management of the information created during a product’s lifecycle is becoming increasingly complicated. Sponsors generally do not have sole responsibility for the creation of information for their product. Due to the nature of the development process, the data will go through multiple iterations, companies and structures before reaching the desk of the regulator. Maintaining the ‘one version of the truth’ is very important as it is these data that support the claims in the label throughout the lifecycle of the product.
The blog discusses the benefits of centralizing the storage of information, either using the sponsors’ systems and processes, or through making use of specialized providers. Over the whole lifecycle of a product, many changes will occur with respect to standards, technology and regulation. Management of the clinical information obtained throughout the conduct of the clinical trials is essential for successful regulatory approval and, potentially, in the defence of a product.
In order to centralize the capture and storage of clinical data in an intelligent way, questions must be asked about the information. Planning the consolidation and traceability of information from an early stage for all studies conducted in house, by different Clinical Research Organisations (CROs), in different standards and across different countries and systems will pay dividends in the end. This blog consolidates current practice and introduces new ideas that are applicable to sponsors and CROs alike. It also introduces the technology aspect, an area that is becoming increasingly important. It is technology that delivers the traceability of the underlying data that the regulators now require.
What is Traceability?
Traceability can be defined in various ways, depending on the environment and process under consideration. Traceability describes the ability to verify the origin, location, or application of an item by means of documented recorded identification. Traceability facilitates transparency, which is an essential component in building confidence in a result or conclusion. (1)
For clinical information, traceability means knowing where your data are located, how information is derived and from which data. At a higher level, it is also what information was used for a decision that led to the label being submitted for approval to the authorities. At the point of submission to a regulatory authority it is important to recognize ‘the one version of the truth’ in support of your label. Centralizing and standardizing the process from your first trials in man through your submission, approval, label extension and marketing phase of the product will also ensure that whenever a question is asked about your label then you can be sure that your response will be fast, traceable and above all, accurate.
Why Is Traceability Important For Clinical Information?
The way in which a company creates, stores and retrieves clinical information has a significant impact on its reputation, efficiency, and ability to keep products on the market. It is therefore essential that the traceability of planned information (ie, clinical trial data) is considered at a very early stage of a product’s clinical development lifecycle.
It is only natural for clinical teams to focus on the information required for submission and approval; whereas the product’s entire clinical lifecycle needs to be considered from a traceability and information storage perspective. At any point in time, authorities may ask for a product’s total trial profile and this is where traceability becomes paramount.
Key considerations are:
- Regulations: regulations governing the conduct and management of clinical trials in a given country. For example, in the United Kingdom (UK) www.legislation.gov.uk/uksi/2004/1031/schedule/12/made Schedule 1 Part 2, 10, states ‘All clinical trial information shall be recorded, handled and stored in a way that allows its accurate reporting, interpretation and verification.’
- Timescales: Many years will pass from first-in-man testing, through submission, approval and marketing, and eventual product death. During this time, there are many occasions when the product’s information may be required for efficacy and safety profiling
- Changing environment: Standards are never standard for long; regulations evolve and technology improves and often becomes more complex. During the product’s lifecycle, a sponsor may change technology provider, collaborate with a new CRO, in-licence, partner or perhaps even merge with another company.
So bearing in mind the considerations above, how can sponsors ensure that in a changing environment, their product’s knowledge store is complete, searchable, traceable and accurate? And what are the benefits that this will bring to the company?
What Exactly Do We Mean By The Traceability Of The Information From a Clinical Development Perspective?
Traceability means that every unique piece of clinical data can be traced through the entire software flow of all relevant application programs, from data capture to final submission documents. Documents and files at any point in the system can be audited for accuracy and completeness against the raw data.
As a starting point, sponsors could ask the question “Where is our clinical information? ”Across a portfolio of studies that may span years from Phase I to Phase 4, across various countries, companies and systems, has there been sufficient upfront planning and management to recognise the one version of the truth at the time it is needed? That is traceability in itself.
At another level, a sponsor may be looking for traceability across the information trail within a study for a particular product. For example, information in the statistical analysis plan (SAP) that also appears in the Clinical Study Report (CSR) and Common Technical Document (CTD) summaries can become traceable and reusable (from a metadata perspective the Clinical Data Interchange Standards Consortium (CDISC) analysis data model (ADaM) is a good example of this). Storing that piece of information once and referencing it from the documents or summary tables provides a centralised, reusable piece of information. Think about where you store one of the most important information points for your study, the p-value. Is it stored in a reusable information store for future use or is it buried in a table generated from your in-house reporting environment and copied numerous times across numerous Word documents?
One final aspect to consider is the decision-making process during the lifecycle of the product. Information and decisions made during the course of a submission need to be traceable back to the raw data. Under the tight timelines during submission, it may not seem efficient to record the reasons for taking a particular approach. These captured gems of information, if preserved for future reference, will save you time, credibility and perhaps your label.
Managing Standards From The Beginning
The biggest problem in managing data is a simple one - time. A product’s lifecycle, from preclinical testing through clinical trials and post-marketing activities, can last for many years. During this time, a sponsor may change technology provider, collaborate with a new CRO, in-licence, partner or perhaps even merge with another company. The clinical data from a single portfolio can therefore end up being stored in multiple locations with different technology providers, utilising different systems and formats.
Technology is now available to manage the vast amounts of information created during a clinical trial and companies and vendors are investing heavily in this area. However, technology alone cannot solve the problem; it is critical to get stakeholder buy-in from the governance and functional teams, to consider what communications and behavioural changes are required and ensure that the process is auditable.
Getting Into the Detail…
Clinical Information Principles
At all stages of the research and development process, the assumption exists that ‘information’ is ‘information’. In general, this paper will use ‘information’ as a term applying to all aspects and all levels. However, it can be useful to think of four different levels of clinical information that contribute to a product’s label:
- Raw data collected during the conduct of the clinical trials
- Summarized and reported, the derived data become Information
- Apply expertise to that Information and it becomes the Knowledge/Conclusions
- That overall knowledge then forms the basis for the label of a product or Wisdom
A label submitted for approval is done so on the basis that the sponsor has proven that a product intended for use in a certain population is safe, is effective in the defined use, and is manufactured to an acceptable quality. A vast amount of information goes into a submission, in the preclinical and clinical research areas, chemistry, manufacturing and controls and of course the regulatory area. In fact, the majority of regulatory knowledge is generated outside of the regulatory affairs function.
In order to be readily available for the right audience at the right time, the information generated from a clinical programme needs to be structured in the correct way. The inclusion of metadata (‘data about data’) to surround the information provides the opportunity to create and maintain the one version of the truth. It also provides the user of the information with a common store referenced from many different types of media, hence gaining efficiencies.
Clinical Data Storage and Traceability
During the conduct of a clinical study, many data points are created using various methods and computer systems.
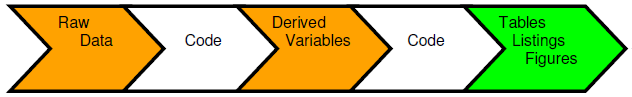
A traditional method of creating usable information is to collect the raw data and input into a data management tool, which is extracted to produce a raw data store for use by the statisticians and programmers. Code is generated to produce derived variables that are stored alongside the raw variables to deliver a reporting database in support of a submission. Further code is then produced that will create the tables, listings and figures (TLFs) that will be used in the first case for the CSRs. A problem with this approach is that these summary tables may contain vital information that are never stored as data points and are only traceable through a cut and paste action or an interface from one environment to another. The output tables and Word documents become additional storage points of the same information. This process is repeated as information is drawn from the CSR into other regulatory documents such as public disclosure reports, Investigator’s Brochures, clinical trial protocols and submission documents. The one version of the truth becomes diluted and spread across multiple documents, formats and locations.
Locating the information point in an information store and referencing it using metadata is an elegant solution to this issue. This approach means that documents do not become the creation and storage mechanisms of information but the users of the information. If information is stored and tracked (tagged) through metadata, then statements in documents can then be traced right back to the raw data. This ability may be critical in certain situations such as in defence of a product label. Working in this way also means that your information also becomes searchable.
Documents should be the users of the information rather than the storage mechanism
Every statement in a product label should be supported by evidence from the raw data that are translated using information, knowledge and wisdom. Documenting and tracking the traceability to this raw data will enable faster access and therefore improve response times to regulatory authorities.
Providing The Constant In A Variable Environment - A New Way Of Thinking?
Many different factors influence the decision-making process when a sponsor considers where to conduct their clinical studies. For example:
- Specific therapeutic experience or knowledge
- History of success
- Access to specific populations
- Location
- Cost effectiveness
- Expertise with certain phases of development
It is unlikely that one unit/CRO will be the optimum choice for every study in a portfolio and therefore studies for any individual compound will continue to be performed across several different units/companies. Since the cost of actually conducting the trial is the largest part of the overall study budget it is understandable that the focus is on this aspect; however, these units/CROs are not necessarily the best choice for housing and reporting the data.
Taking the approach of including the data collection, storage and reporting for a study with a full service CRO means that programme information becomes scattered across various companies and systems using a variation on a standard with little or no traceability. This compromises on the quality of the input and output from a biometrics perspective and loses the vital consistency required at a later stage of the product’s lifecycle.
Why Not Make The Information Traceability The Constant?
You could have a solution provider to supply not only the technology and the standardisation but also, and most importantly, the knowledge of the clinical trial process and therapy area. Consider the advantages of having the same medical writer provide input to your phase 1 protocol, prepare your CSR, and eventually the publications that are created for the marketing of the product. The saving in terms of the reuse of intellectual property across a brand is obvious from a data management perspective and output generation perspective. Combine that with the consultancy input from a knowledgeable statistician across the development programme - your studies can be designed so that the data fit together seamlessly if required for submission.
Think of it as your bank of information. When selecting a bank to support your business you will probably look for one that can provide you with a secure place for your money with accessible interfaces to work on a global scale, ie, one that will turn your foreign currency transactions into your currency of choice. They will help you do business with many different companies but always provide you with the applications and interfaces to standardise your money and give you the ability to pay out in different currencies to your customers. A centralised biometrics organisation will do the same for you with your clinical information. With your data collected from all sources - your own company, partners such as CROs, laboratories, research institutes - and over the lifecycle of a product, a centralised clinical data specialist will be able to supply you with the information you need, in the right format, at the right time and to different regulatory customers.
Generating The Traceability Up Front
Creating a standard to follow can be difficult to do within an organisation but maintaining it is even harder. The introduction of standards such as CDISC has given companies major challenges to implement and uphold. Standards then need to be maintained through new versions and the task of traceability becomes progressively more complicated. Systems driven by metadata are becoming increasingly important to deliver long-term benefits. Metadata control the way that standards are managed and the traceability between your data and information. Using these types of tools provide great advantages in terms of traceability although they do require maintenance and governance which can be resource heavy.
Traceability of an information point can start to make use of such environments. It is possible to know the data used in the generation of such information when a query is raised. The one version of the truth is found with confidence and in a timely manner.
Through some environments it is possible to use a reverse impact analysis across a mapped workflow to show visually how a summarised piece of information was generated, from what data points and using which algorithms. Managing this internally as part of your ongoing submission timescales can be daunting if you are using multiple partners. It is possible if you are relying on one CRO to manage your data. Centralising this process becomes your constant. Customers of the data are provided with access to the information they need, when they need it. This model also reduces your technology burden internally and compliance with 21CFR Part 11 is all handled for you.
There are many different systems offering such functionality. Sponsors spend a great deal of time and effort choosing the environment and then need to implement and maintain the system. They also have to re-train or recruit resources to manage the process. All of this happens during the same time they are delivering to the requirements of the clinical trials. Companies that provide access to the best-of-breed industry tools and have trained resources for these environments will obtain great benefits.
Conclusion
Centralising clinical data services provides sponsors with a constant within their clinical trial submission process. The traceability of that information over time is managed from Day 1 of a product’s lifecycle and information points along the course of time are stored once, progressed to stay in-line with the evolution of the standard, and are always accessible. When questions are asked about the product, the responses are managed through the information trail that has been generated and sponsors can be reassured that they have the one version of the truth.
Every life science company faces the same challenges in the management of their clinical information. To be more effective in your management of clinical knowledge and information it is important to consider the basic principles of how data, information, knowledge and wisdom are generated and how these can traced throughout the lifecycle of a product. Additionally knowing how information flows across and within functions is important to gain a true understanding of what information is in place to support the label of a product and the speed and accuracy at which it will be accessed.
The ultimate metrics around how good we are in terms of the submission, relate to the approval timeframe and the quality of the submission. In essence, if we can provide data of such quality that a question is not raised, then our response times are kept to a minimum and this is a true measure of the quality of information supporting the statements in the label.
References
(1) CDISC Analysis Data Model (ADaM) Team, 2009 “ CDISC Analysis Data Model, Version
2.1” available on CDISC website at http://www.cdisc.org/standards
Subscribe to the Blog
FOLLOW QUANTICATE ON:
© 2024 Quanticate