
This article has been updated in May 2020 to reflect the guidance by European Medicines Agency and other regulatory bodies on remote monitoring during the coronavirus disease (COVID-19) pandemic.
Within the last few decades the number and complexity of clinical trials has increased considerably, not only across the industry but also within individual companies. With this increase comes the enhanced pressure of effectively monitoring these trials.
Sponsors and regulators appreciate that there is no “one size fits all” when it comes to monitoring plans; the Food and Drug Administration (FDA) and European Medicines Agency (EMA) are encouraging sponsors to explore more effective monitoring strategies, promoting a more risk based approach for the monitoring of clinical trials, especially with the changes in ICH GCP E6 R2 Addendum in November 2016.
Moving away from 100% Source Data Verification
A variety of approaches can be used by sponsors to ensure monitoring obligations are fulfilled. Traditionally, this has involved on-site visits where monitors are required to check a high percentage of the data entered on the case report forms (CRFs) against source, to ensure that the protocol is deployed correctly, and the trial is conducted as per Good Clinical Practices and applicable regulatory requirements. This has often involved 100% source data verification (SDV). The size, complexity and number of modern clinical trials mean that complete on-site monitoring is becoming an ineffective, expensive and inefficient process. Sponsors sending clinical research associates (CRAs) to sites ,to perform 100% SDV, results in large travel costs that a pharmaceutical company has to add to its budget or a full service Clinical Research Organization (CRO) adds on as a pass-through cost.
Effective trial monitoring is critical for the protection of subjects and the integrity of the data produced, but where on-site monitoring was once a requirement by regulatory bodies for critical study parameters, there is now an acceptance to move away from 100% SDV, as regulatory bodies understand that sponsors need to look for efficiencies in the cost of drug development, and research shows that 100% SDV often fails to pick up all issues. Remote monitoring is seen as an acceptable addition to the process. In a remote monitoring approach, combined with Risk Based Monitoring, you are moving away from 100% SDV on-site. The new process involves remote source data verification (rSDV) where the verification of source data is done remotely via a centralized location, by a specialist team. It is possible for multiple site’s source data to be checked at a single centralization location instead of going to a particular site. This introduces us to the concept of centralized monitoring.
When centralized monitoring and an rSDV approach is considered, the focus is usually on certain sites such as; high recruiting sites, low compliance sites, and sites with predicted or previous protocol deviations/violations. Also it is common for centralized monitoring and rSDV to focus on data points related to primary end points, safety data and certain trial related processes.
Remote Source Data Verification
rSDV can be conducted using the below options:
- Site teams sharing redacted copies of trial related source documents with the centralized team/monitor, using mobile apps or other technologies like uploading scanned PDFs to validated portals with audit trails.
- Video review of records; site staff sharing their computer screen using a secure video conference application.
- Providing restricted remote access (read only) to trial participants’ electronic medical records (EMR).

Centralized Monitoring
Once a single centralized team is responsible for the SDV and the Data Quality Oversight, it becomes possible to identify trends and potential problems that go beyond transcription problems from source documents. A single CRA checking a single site may observe that patient data are consistent and properly transcribed from the patient records to the CRF. Only the Centralized Remote Monitors can spot the critical fact that one site is producing completely different data to the others through, e.g., a mis-calibration. This permits problems to be identified and resolved in real time, dramatically reducing costs and problems when issues are only discovered post-data lock as in the past[1].
To use remote monitoring effectively, a risk assessment must be performed and included in the monitoring plan prior to the start of any clinical program and adapted as the program evolves.
A monitoring risk assessment is required to ensure:
- Adequate protection of the rights, welfare and safety of the subjects.
- Quality and integrity of the data provided by the sites.
The risk assessment should be used to define the required level of on-site and remote monitoring:
- On-site monitoring should concentrate on those aspects of the study defined in the risk plan as critical to the study integrity (for example, informed consent, endpoints that are interpretative or subjective, drug accountability/randomization integrity, SDV, site non-compliance, trends in under reporting of adverse events (AEs) or more serious adverse events (SAEs).
-
Remote monitoring should concentrate on those activities which can be reviewed and monitored remotely, such as consistency checking, data completeness, identification of sites with high error rates or protocol deviations/violations. Also, where data are acquired by electronic data capture systems, e.g., central laboratory, electro-encephalogram (ECG)/imaging data, electronic patient-related outcomes (ePROs), or interactive web response (IWRS).
The monitoring plan should retain flexibility to increase on-site monitoring in case of issues or problems, e.g., sites with a high number of protocol deviations/violations or poor data quality may require additional visits to re-train and mentor study staff, or conversely sites with low AE rates contradictory to predictions or other site metrics. To facilitate this, tolerance levels for data collection, protocol deviations/violations, AE reporting etc., must be set at the outset and automatically trigger increased site visits when missed.
In the past, the process of determining ‘poor data quality’ at a site, so that an on-site monitor is potentially triggered, has been known as central data analytics (CDA). Since more technology vendors have developed software with visualizations that use statistical algorithms to analyze subject and site data, to detect erroneous/fraudulent data anomalies, or outliers, the process has become known as Data Quality Oversight. These Data Quality Oversight checks can be applied to any trials regardless of the monitoring preference.

Remote Monitoring and Remote Source Data Verification (rSDV) During the Pandemic
The COVID-19 pandemic caused an increase in the demand for remote monitoring and RSDV. Health care professionals faced difficulties in carrying out their duties, for example, social distancing, travel restrictions, and lockdown conditions prevented CRA visits and patient participation in clinical trials. Also, the integrity of clinical trials was affected with the increased potential for missing data, study conduct issues and general noncompliance. Data Quality Oversight and Centralized Statistical Monitoring (CSM) are well suited to help investigate these issues.
European Medical Agency Stance
In response to the restrictions and the consequent impact on the conduct of clinical trials, the EMA has proposed methodologies for clinical trial monitoring which are flexible and harmonized to comply with the integrity of trial related data, protect trial participants and the safety of health care professionals[2].
The EMA suggests the below monitoring methodologies instead of on-site monitoring visits:
- Centralized monitoring and central review of data collected
- Off-site monitoring
- Remote source data verifications (rSDV) as long as redaction does not burden sites unnecessarily
- Any combination of above monitoring approach
FDA stance
The “FDA Guidance on Conduct of Clinical Trials of Medical Products during COVID-19 Pandemic” states that if on-site visits are no longer feasible then trial sponsors should consider the use of centralized or remote monitoring approaches to maintain oversight. US guidelines do not specifically detail rSDV because they are not directly affected by the EU’s data privacy laws which EMA member states need to abide by.
Increased Interest in Remote and Centralized Monitoring
Sponsors have to decide which approach is suitable for their clinical trials to protect the rights, safety, and wellbeing of trial participants along with the protection of clinical trial staff at site.
On-site monitoring is widely used and accepted for all the stakeholders of clinical research and regulatory authorities but it is well understood that traditional on-site monitoring may no longer be the most practical or safest form of monitoring. The COVID-19 pandemic has accelerated interest in the remote monitoring approach[3].
This demand can also been evidenced by the increase in traffic to this article. Traffic was consistent for multiple years, but at the start of the global pandemic the search volume traffic for 'remote monitoring clinical trials' has increased and reflected in the graph below.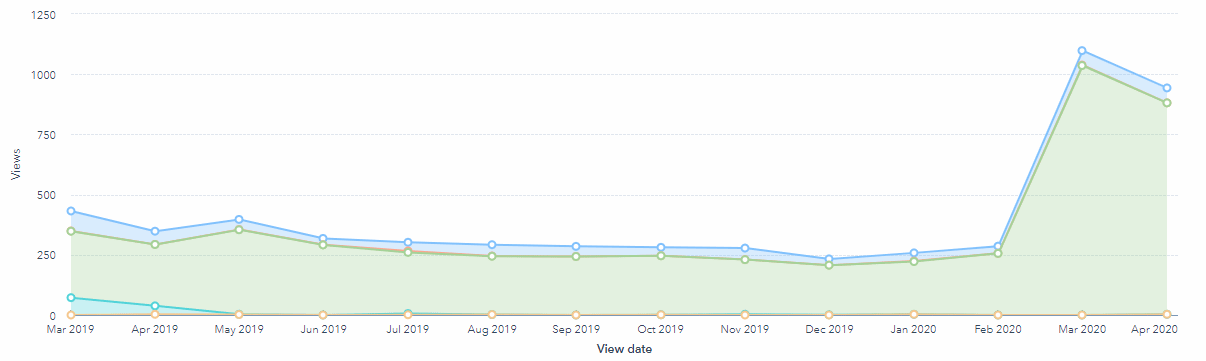
How to perform rSDV for a Marketing Authorization Application Approval in the Current Pandemic?
For sponsors submitting trials for Marketing Authorization Application (MAA) approval to the EMA, there is a workaround to use remote monitoring and rSDV that takes into consideration the EMA's concerns around redaction.
The recent guidance released by EMA on the management of clinical trials during the COVID-19 pandemic allows rSDV for trials, if necessary. All decisions to adjust clinical trial conduct including monitoring should be based on a risk assessment by the sponsor.
In the case of the informed consent form (ICF) being updated to allow transfer of redacted data to a central review team, the below options for ICFs can be considered by the sponsor:
Option #1: A separate single-page ICF can be executed. The signed ICF can be uploaded in an app.
Option #2: A checklist with the trial participant’s signature can be taken by the site. The signed checklist can be uploaded into an app, as it will only contain subject initials, screening number, and site number.
There are a few proposals on redacting the data while/after sending it to the centralized team.
- An independent team/person who is not part of study or CRO/sponsor should perform redaction.
- There should be a technology in place where subject details (within the header of the document) can be redacted by an automatic program while uploading to an app by the site team.
The EMA released guidance for remote monitoring on data protection, monitor responsibilities, subject confidentiality, etc. The guidance helps a sponsor/CRO to take the decision on a remote monitoring approach.
Quanticate offers Data Quality Oversight (DQO) of site data which uses statistical analytics to generate reports to improve data integrity as outliers and data anomalies are discovered. DQO can be applied to any clinical monitoring method. Quanticate can also provide consultancy around rSDV and methodologies to conduct centralized remote monitoring and centralized statistical monitoring. Request a consultation below if you would like to hear how we could improve data quality or monitoring efficiency and a member of our team will be in touch with you shortly.
References
1) http://journals.sagepub.com/doi/full/10.1177/2168479014554400 accessed 03/09/2018
Related Blogs:
- The Rise of Risk Based Monitoring in Clinical Trials
- The Rise of Risk Based Monitoring [Infographic]
- The Evolution of Risk-Based and Remote Monitoring
- ICHGCP E6 Addendum R2 - What do you need to know?
This blog was originally published in 13/01/2011 and updated on more than one occasion as new regulatory guidance and industry trends emerge on monitoring techniques.



